LC/ESI/HRMS非靶向分析中的定量方法:实验室间比较

题目:Quantification Approaches in Non-Target LC/ESI/HRMS Analysis: An Interlaboratory Comparison
摘 要
利用液相色谱电喷雾电离高分辨率质谱(LC/ESI/HRMS)的非靶向筛查(NTS)越来越多地用于鉴定环境污染物。ESI/HRMS中化合物电离效率的差异,导致响应差异很大,并使定量分析复杂化。尽管NTS中没有真实标准的定量方法越来越多,但这些方法是在有限且多样化的数据集上进行评估的,这些数据集的化学覆盖范围不同,在不同仪器上收集,使无偏倚性的实现比较复杂。在由NORMAN Network组织的实验室间比较中,我们评估了来自37个实验室的41种NTS方法的5种定量方法的准确性和性能变异性。三种方法基于替代标准定量(母体转化产物、结构相似或接近洗脱时间),两种方法基于预测的电离效率(RandFor-IE和MLR-IE)。实验室使用内部NTS工作流程分析了加标45种化合物2种浓度水平的HPLC级水、自来水和地表水,以及在6个已知浓度水平的41种标准品。通过比较定量方法、仪器和实验室的估计浓度和加标浓度来评估方法的准确性。RandFor-IE方法表现最佳,报告的平均预测误差为15×,超过83% 的化合物定量误差在10×误差内。尽管仪器和工作流程不同,但不同实验室的性能是稳定的,并且不取决于水基质的复杂性。
研究背景
液相色谱电喷雾电离高分辨率质谱(LC/ESI/HRMS)是最常用的水分析技术之一。HRMS的灵敏度、准确性和分离能力的提高使大量极性和半极性有机微污染物的鉴定成为可能。由于水样中存在大量污染物,分析方法已从靶向转向疑似和非靶向方法。样品不是靶向特定化合物,而是筛选所有检测到的质荷比(m/z)。目的是在不使用分析标准品的情况下,初步鉴定疑似列表匹配或所有检测到的特征(独特的保留时间(RT)和准确的m/z)。尽管如此,最终需要购买和分析标准品才能进行明确的验证。然而,由于分析一个样品可以产生数以万计的检测特征,因此即使不是不可能,也无法获得所有初步鉴定化合物的参考标准品。
尽管LC/ESI/HRMS是目前首选的分析技术,但定量本身就受到限制。ESI中的电离效率(IE)在很大程度上取决于化合物的物理化学性质,例如极性、酸碱特性、分子体积、所用洗脱液的性质和电离源几何结构。因此,化合物的IE可以相差几个数量级。因此,从LC/ESI/HRMS分析获得的信号并不能指示样品中化合物的绝对浓度。定量信息可以通过标准曲线法获得,在完全鉴定之前仍然无法获得。因此,在过去十年中,已经开发了几种无需分析标准品即可定量LC/ESI/HRMS NTS检测到的化合物的方法。一些方法使用替代标准品(结构相似或具有相似的色谱行为)进行定量,而其他组织则依靠机器学习来预测检测到的化合物的IE,然后应用预测的IE进行定量。最近,这些方法在杀虫剂、药物及其TP上进行了评估,发现基于IE的预测模型提供了最准确的结果。该比较基于在一个实验室的一台仪器上分析的样品。因此,目前尚不清楚定量方法的准确性在多大程度上取决于仪器和/或所使用的处理软件,或分析人员的经验。
在这项针对欧洲、北美和澳大利亚的37个实验室的NORMAN实验室间研究中,测试和评估了五种没有分析标准品的定量方法。具体来说,比较了三种替代标准定量的方法和两种基于预测电离效率的方法。每个参与实验室使用其标准非靶向LC/ESI/HRMS工作流程分析了15个样品,同时提供了一份加标化合物的疑似清单。这些样品由三种加标45种化合物的水基质组成,包括两种浓度水平的工业化学品、农用化学品、食品添加剂、药物、个人护理产品和天然产物。参与者不知道加标化合物的浓度。此外,将6个已知浓度的41种化合物的超纯水和3种空白基质中的标准溶液运送给所有参与者。该研究旨在(1)比较不同实验室中五种定量方法的性能和准确性的变异性;(2)评估不同仪器对他们分析的影响。
主要发现
1. 化合物选择和稳定性评估
化合物的选择基于环境相关性,重点是水,旨在涵盖广泛的化学空间。这包括具有不同极性和电离电位的化合物,以及具有已知TP的化合物。化合物选自NORMAN SusDat并基于文献中的信息。图1a显示了基于响应因子(RF)和保留时间的化合物分布,而图1b显示了一个实验室中标准化合物和疑似化合物的RF分布。每种化合物的RF计算为峰面积与浓度的比值。
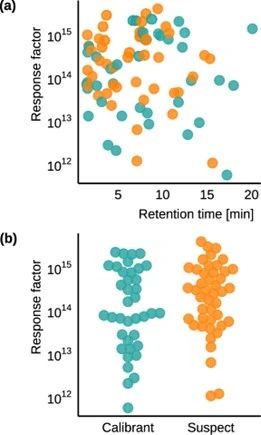
图1 加标到水样中的选定化合物的分布:(a)保留时间范围和化合物响应因子在25 min梯度中的分布,以及(b)标准化合物和疑似化合物的响应因子分布。
2. 参与者报告的浓度估计值
对所有参与者报告浓度的分析揭示了一些趋势。基于电离效率的方法通常比基于替代标准的方法产生更好的定量准确性。对于这两种方法,样品和数据集中的大多数化合物的误差都在10倍以内,但也有一些例外。MLR-IE方法来自两个数据集的结果比其他数据集的偏差更大(图2a)。然而,接近洗脱方法产生的总体误差最高,高达约1,000,000×。尽管使用了不同的工具,但在数据集中可以看到类似的总体趋势。对数误差的分析(图2c)表明,大多数方法更容易出现低估的情况,尤其是parent-TP和结构相似性方法。基于电离效率的方法和接近洗脱方法不太容易出现低估。然而,基于电离效率的方法对几乎所有异常值都被低估了。低估是不可取的,因为一致的低估可能会导致忽略环境/生态毒理学相关浓度下的化合物。相反,对于定量估计,最好使用轻微的高估,以确保包括所有可能的有害化合物以供进一步调查,尤其是对于环境分析和风险评估。
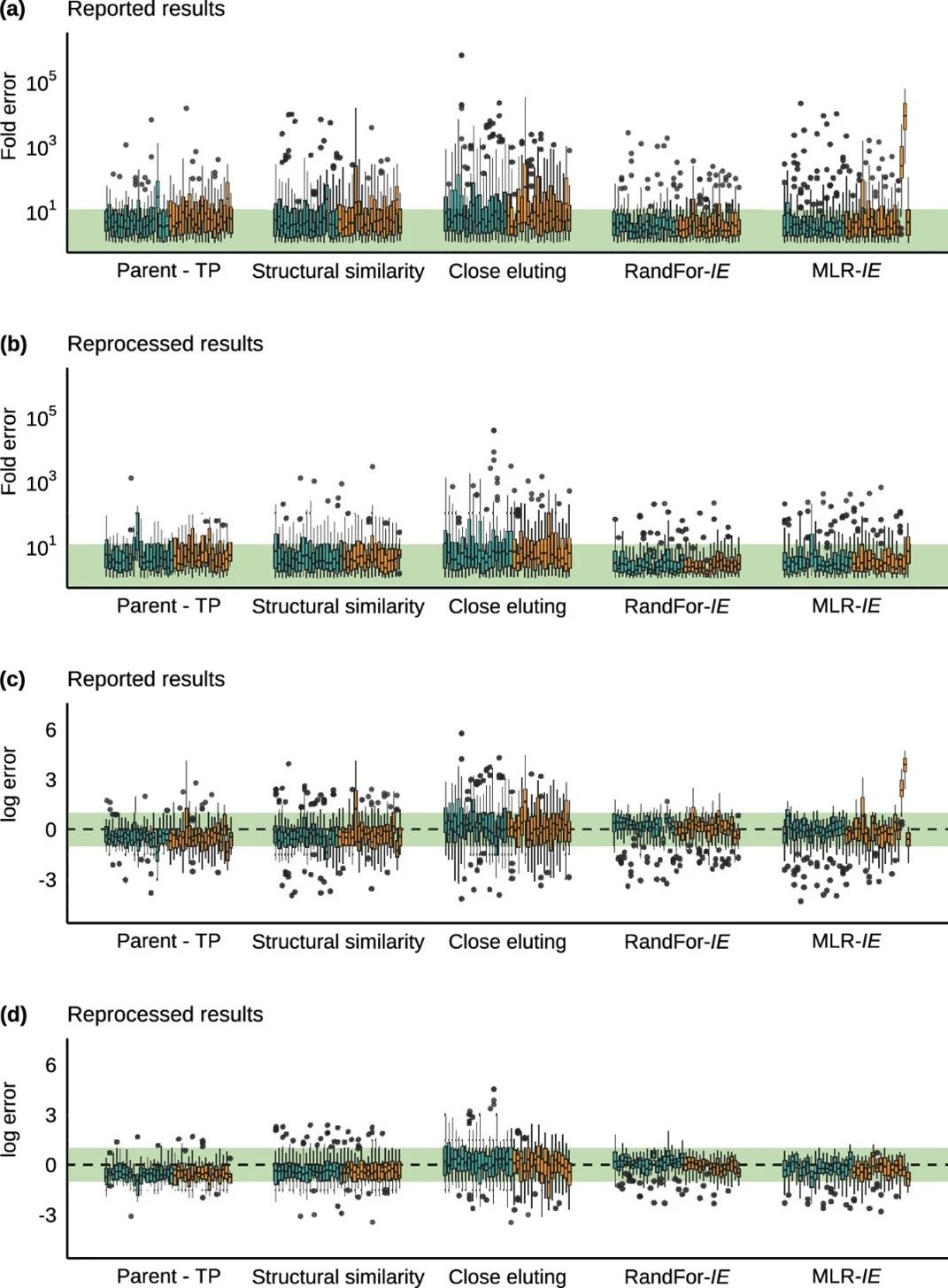
图2 样品s1a(HPLC水中的高浓度样品)数据集上每种定量方法的预测误差。绿色区域显示10倍误差和等效对数误差。蓝色框来自orbitrap HRMS分析,橙色框来自ToF HRMS分析。折叠误差显示在(a)中,相应的对数误差在(c)中。(b)(d)显示了再处理数据的相应图表。
观察到在不同基质和浓度水平中获得的峰面积之间存在统计学上的显著差异(调整后的p < 0.05)。然而,只观察到对总体结果的轻微影响,因为预测误差在样本中基本一致。显著但非系统性的差异表明,基质效应在很大程度上取决于样品和色谱的特定组合。此外,对于本研究中的水样,基质效应的影响似乎小于定量方法的不准确性。使用Friedman检验比较数据集的平均折叠误差,并观察到所有样本的统计学显着差异(p < 0.05)。同样,所有样本中定量方法的平均折叠误差在统计学上也不同。
3. 离群值分析
为了评估经常与高误差相关的化合物的趋势,研究了箱须图(图2)中的异常值。但是,由于数据集之间的差异,在一个数据集中属于异常值的化合物可能会在另一个数据集中产生较高但非异常的误差。另一方面,只有少数异常值在10×误差范围内(对应于−1至1的对数误差),并且这些异常值分析未考虑在内。由于分析涉及两个误差指标(折叠误差和对数误差)、五种量化方法、六个样本和40个数据集,因此根据每个数据集中的检测频率,每种化合物最多可以被确定为2400次出现的异常值。尽管没有看到明显的趋势,但一些化合物在多个数据集中表现为异常值,或者在更少的数据集中出现的频率更高。例如,灭多威、阿特拉津去乙基二异丙基、苏丹红I、毒死蜱和苯并噻唑在数量最多的数据集中作为异常值出现;分别有22个(55%)、22个(55%)、20个(50%)、19个(47.5%)和18个(45%)数据集。同样,灭多威、丁胺、克霉唑、萘普生和毒死蜱是最常见的异常值,分别出现98、41、57、49和58次,考虑到检出率,异常值率分别为7.8%、5.9、5.4、5.3和4.2%。
这些异常值都属于不同的化合物类别,离子质量范围为74至350 Da,RT范围从非常早洗脱到极晚洗脱。因此,对于为什么这些特定化合物经常出现异常值,无法得出一般性结论。相反,我们检查了每个样品中高加标和低加标之间的峰面积比,以查看具有偏差比值的化合物是否也产生了最高的误差,因此会被视为异常值。比率超出范围的化合物通常不是最常见的异常值,反之亦然。有时,比率超出范围的化合物甚至不被视为异常值,也没有特别高的预测误差。
4. 定量方法对预测误差的影响
定量方法观察到的高预测误差可能有多种原因,并且这些原因可能因方法而异。对于除接近洗脱法之外的所有方法,定量都需要一个暂定结构。因此,这些误差可能部分归因于化合物注释的不确定性。在这里,由于提供了疑似列表,此错误源受到限制,旨在对方法本身进行更公平的评估。然而,在现实中,这是一个真正的问题,可能会增加错误,特别是对于不正确或不确定的注释。
替代标准品与可疑/未知物之间的响应差异较大可能会导致更高的误差。例如,TP的疏水性通常低于其母体化合物,从而导致电离效率降低。这可以解释parent-TP方法中出现的普遍低估。这也影响了本研究中结构相似性方法的预测误差,因为大部分加标化合物是TP,而它们最相似的替代标准品主要是各自的母体化学品。但是,有时另一种校准化合物在结构上比母体化合物更相似。对于两种苯并噻唑类化合物,最相似的化合物是苯并三唑,而对于阿特拉津TPs,西马津比它们的母体更相似,而对于二甲双胍,咖啡因在结构上比鸟苷类更相似。这些TP的RF通常与结构最相似化合物的RF更相似,而不是与其各自母体化合物的响应因子的RF更相似。尽管如此,仍观察到了近2个数量级的差异。对于这些TP,与parent-TP方法相比,结构相似性方法的平均预测误差有所改善,这表明如果TP的结构与其母体化合物非常不同,则结构相似的化学品可能更适合定量。
对于本研究中包括的疑似化合物,除2-氨基苯并噻唑、二甲双胍、灭多威、莫诺隆、萘普生和噻苯达唑外,大多数化合物的RF低于其结构最相似或母体化合物。毒死蜱及其最相似的标准品异丙甲草胺的RF差异最大,为3个数量级。然而,仅凭疑似化合物和标准化合物之间RF的巨大差异并不能解释最高的绝对误差。例如,阿特拉津-去乙基-去异丙基和阿特拉津之间的RF差异与阿特拉津-去乙基-去异丙基-2-羟基和阿特拉津之间的差异大致相同,但平均倍数误差却大不相同。关于接近洗脱方法,RT可能会根据分析中使用的色谱条件而发生很大变化。因此,在不同液相色谱条件下,最接近的洗脱化合物的分配因数据集而异。在本研究中,虽然主要使用反相色谱,但色谱柱尺寸、粒径和固定相,以及流动相组成、添加剂和pH值因分析而异。因此,在数据集中,不同的标准化合物被指定为最接近同一疑似化合物的洗脱化合物,这表明这种方法的不稳定性。接近洗脱分配从二甲双胍的4种标准化合物到异丙甲草胺-ESA的16 种不同标准化合物不等。然而,不同实验室之间接近洗脱分配的波动似乎对预测误差没有太大影响。例如,异丙甲草胺-ESA具有16种不同的紧密洗脱分配,其平均倍数误差和标准偏差低于二甲双胍和阿特拉津-去异丙基。
对于基于电离效率的方法,预计模型训练的化学空间之外的化合物的误差会更高。虽然用于MLR-IE方法的在线应用程序提供了有关模型是否覆盖疑似化合物的信息,但RandFor-IE方法没有,也没有提供有关所用标准化合物的信息。因此,对本研究中使用的疑似化合物和标准化合物,以及电离效率模型中使用的训练化合物和NORMAN SusDat中的LC/ESI(+)适配化合物进行了主成分分析(PCA)。从图3中的前两个组分可以看出,除了少数标准化合物外,本研究中使用的大多数化合物似乎都被两个模型的化学空间所覆盖。疑似化合物利血平和丁胺似乎没有被模型覆盖,因为它们位于被覆盖的化学空间的边缘。在基于IE的方法中,利血平既没有最高的预测误差,也没有作为异常值出现的次数最多,而丁胺是两种方法的多个数据集中误差最高的五种化合物之一。此外,对数IE到对数RF值的不完美变换也可能导致更高的误差,但是,这里没有对此进行研究。
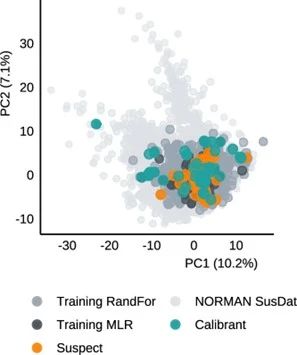
图3 本研究中标准品和疑似物,以及基于IE的模型的训练化合物和来自NORMAN SusDat数据的PCA(前两个组分)
建模方法的结果可能难以解释,尤其是在置信度、可靠性和不确定性方面。NTS社区正在努力将不确定性估计纳入定量分析中,以及为基于IE的模型的估计添加置信度信息,研究小组正在开发中。例如,MLR-IE为预测浓度提供了一个范围,RandFor-IE现在以正模式给出估计值的置信度。然而,在试验时尚不可用。这些努力可以更自信地解释结果。
5. 数据质量对预测误差的影响
除了定量方法如何影响预测误差外,数据的质量还可能影响误差的大小。例如,无论采用何种定量方法,由于化合物识别错误、质谱噪声或处理过程中的其他问题而导致的化合物积分错误也可能导致更高的预测误差。此外,源裂解和加合物形成会显著影响化合物的RF,具体取决于其性质。因此,建议使用加合物和源内片段的峰之和进行定量。在本研究中,一些已知的加合物和源内片段包含在提供给参与者的嫌疑人列表中;然而,在研究之前并未对此进行广泛调查。此外,此处包含的基于IE的方法只能应用于质子化物种,因此,包含其他加合物不会影响这两种方法。
对MLR-IE方法误差较高的两个数据集进行了调查,以了解偏差结果的来源。最初,其中一个数据集不包括MLR-IE方法的报告结果,但在后期使用报告的峰面积计算,没有对校准曲线进行质量评估。这导致在MLR-IE方法的协调步骤(将对数IE变换到对数RF)中使用了多个负斜率,这可能对结果产生了负面影响。对于第二个数据集,协调步骤中不包含负斜率。然而,研究发现,对于这两个数据集,与较高浓度的样品相比,MLR-IE方法最后一步中使用的超过60%的归一化峰面积在较低浓度的样品中更大或相似。然而,两个数据集的非归一化峰面积显示出预期的模式,浓度越高的样品峰面积越高。事实上,在其中一个数据集中,阿特拉津-d5浓度较高的样品峰面积比浓度低的样品高约10倍。这是疑似化合物的预期模式,但不是同位素标记标准品的预期模式,因为它们在所有样品中都以相同的浓度水平加标。这种差异可能是MLR-IE方法观察到异常值错误的原因。
此外,一些化合物在某些数据集和方法中具有很高的误差。在接近洗脱方法中从一个数据集中提取西玛津-2-羟基和2-氨基苯并噻唑,在parent-TP和结构相似性方法中从另一个数据集提取阿特拉津去乙基二异丙基。关于西马嗪-2-羟基和2-氨基苯并噻唑,在所讨论的数据集中使用相同的标准品,即丁酮威(质子化物种)进行了定量。在该数据集中,还报道了丁酮威的铵和钠加合物。然而,报道的[M+H]+和[M+NH4]+和[M+Na]+加合物的RT差异很大(4分钟),加合物具有相同的保留时间。因此,有理由相信,西马嗪-2-羟基和2-氨基苯并噻唑的高误差是由于该特定数据集中的错误分配问题造成的。关于阿特拉津去乙基二异丙基,对于相关数据集,仅在一个样本中检测到,且仅在一次重复中检测到。因此,来自该数据集中阿特拉津脱乙基二异丙基的信号可能是伪影,而不是真实信号,这可以解释高误差。这些数据集中的例子表明了数据质量(包括结构分配)对于准确量化的重要性。
6. 基于再处理的原始数据的浓度估计
对参与者的原始数据进行再处理,并重新计算浓度,以使用一种一致的方法和操作人员评估报告结果中观察到的趋势与再处理结果的相关性。由于峰的再处理和最终评估是由一名专家对特定数据完成的,因此该工作流程比疑似或非靶向筛查更具针对性。尽管如此,由此产生的预测误差揭示了与报告结果相似的趋势,基于电离效率的方法的结果更准确,而目前的大多数方法都存在低估问题,如图2b,d所示。MLR-IE方法的平均折叠误差从3000×降低到11×,RandFor-IE方法的平均折叠误差从15×降低到5.4×。尽管与其他指标相比,中位倍数误差的改善不那么明显,但根据Wilcoxon符号秩检验,接近洗脱、RandFor-IE和MLR-IE方法的差异仍然显著。对于parent-TP和结构相似性方法,结果没有显著改善。如图2b,d所示,MLR-IE方法中偏差结果最大的两个数据集在报告结果中的误差与再处理数据中的其他数据集处于同一范围内,这有助于该方法的巨大改进。
准确性的提高表明,数据处理及其质量控制需要在嫌疑人筛查和NTS中统一。建议在NTS中以不同稀释度运行样品,原因如下:确保在线性范围内进行分析以及评估基质效应。此外,由于稀释液之间的RT对齐,多重稀释液的分析可能会减少仪器伪影的发生。同样,纳入更多的加合物可能有助于进一步提高鉴定的可信度,因为一次分析中的所有加合物都应该具有相同的RT。
7. 结论
对五种常用定量方法的比较表明,基于电离效率建模的方法总体上优于替代标准方法,尤其是在考虑10×误差以内的化合物定量百分比时。RandFor-IE方法在所有评估点的报告和再处理结果中均得出最准确的浓度估计值,而接近洗脱方法在两组结果中产生的预测误差最高。MLR-IE方法在再处理结果中产生了仅次于RandFor-IE方法的第二准确浓度估计值。对于超过83%的化合物,两种基于IE的方法都提供了10×以内的估计浓度,这对于定量非靶向筛查来说被认为是可接受的准确性。然而,定量方法仅限于标准化合物的化学空间或训练用于IE预测的模型时使用的化合物。虽然对于本研究中使用的化合物来说不是问题,但应谨慎考虑适用性域外化合物的结果,以确保其准确性。此外,在测试的方法中,只有接近洗脱法可用于定量没有暂定结构的化合物,与这里的其他方法相比,这是一个优势。
尽管使用了不同的工具和工作流程,但在不同的数据集中观察到了类似的趋势。这表明,虽然工具效应可以影响预测误差的总体幅度,但定量方法之间的相对误差保持一致。在再处理的结果中,与报告的结果相比,预测误差更小;然而,观察到了相同的总体趋势。这凸显了跨不同工作流程进行统一数据处理和质量控制的必要性,但也凸显了定量方法固有的不准确性,这些不准确性不会受到数据质量的影响。重要的是,这些误差以样品中存在正确的结构为前提。在现实中,与鉴定相关的不确定性可能会传播到定量步骤,从而增加预测误差。
根据本研究中提供的结果,在首先仔细评估所获得数据的质量后,建议尽可能使用基于电离效率的定量。为了尽量减少与定量相关的误差,我们强烈建议遵循NTS的一般推荐指南,尤其是数据处理。
原文链接:
https://doi.org/10.1021/acs.analchem.4c02902
说明:本推送只用于学术交流,如有侵权,请联系删除。
投稿&合作请联系:ecs_pku@163.com